
Clone Screening
Dedicated solutions for clone screening and single-cell isolation, increasing the probability of identifying rare, high-value clones using advanced imaging, automation, and precision robotics.

Clone screening solutions for monoclonality verification, colony picking, and single-cell isolation
Our clone screening solutions enable researchers to confidently verify monoclonality, efficiently pick colonies, and automate workflows to streamline cell line development. With precise image-based documentation, automation, and advanced robotics, these systems provide scalable, efficient solutions for antibody discovery and synthetic biology.

Clone Screening

Designed to improve the probability of finding rare clones and ensure objective selection of mammalian and microbial colonies. Our proven techniques establish true monoclonality and automated colony picking, accelerating your projects with reliable results.
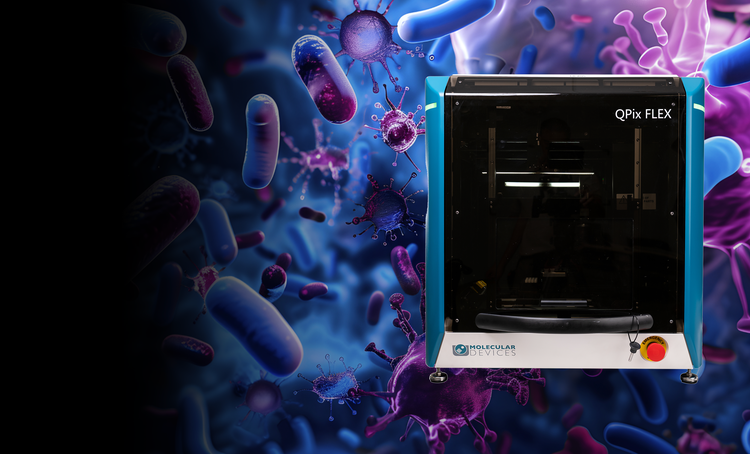
Microbial colony screening and selection
The QPix® Series Microbial Colony Pickers automate synthetic biology workflows, enabling fast, precise selection and screening of microbial colonies. These systems are ideal for applications such as DNA assembly, protein engineering, and antibody discovery, offering scalability and efficiency.
Mammalian colony screening and selection

The ClonePix 2 Mammalian Colony Picker is designed for mammalian cell line development, streamlining the process from initial clone screening to monoclonal verification. These tools ensure accuracy and efficiency in antibody discovery and biopharmaceutical development.
Single-cell cloning
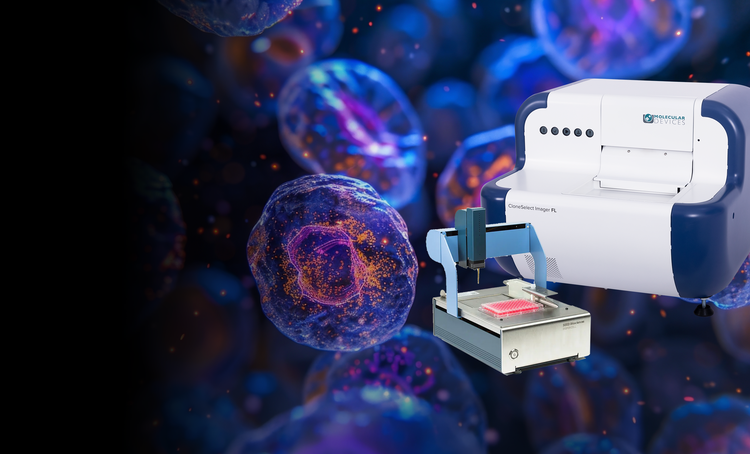
Our single-cell analysis systems enable the efficient isolation of viable single cells, providing increased cloning efficiency and reducing contamination risks. These solutions are essential for workflows requiring proof of monoclonality and accurate cell tracking.

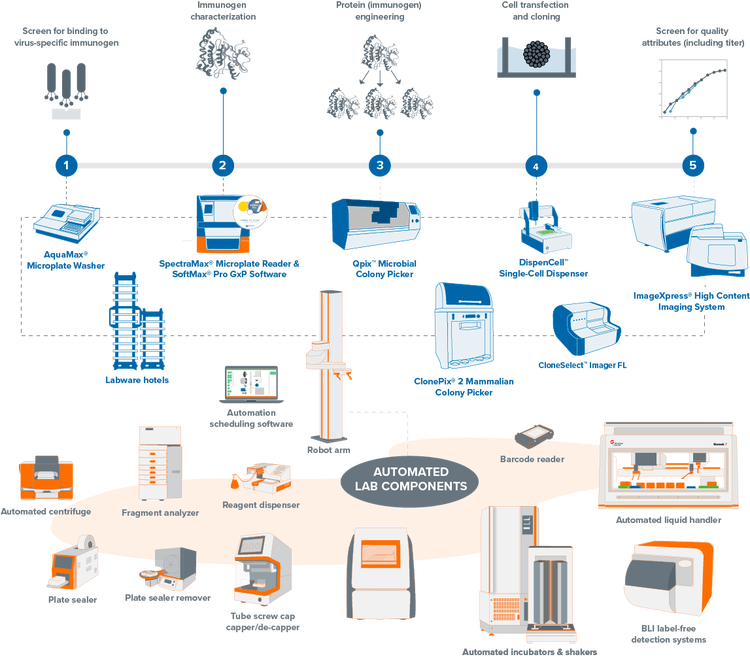
Automated, high-throughput clone screening
Fully-integrated, lab automation solution for molecular cloning, antibody discovery, and monoclonality. Our automated clone screening workflows integrate laboratory devices to increase your throughput and efficiency while reducing human interaction.
Transform your clone screening with automation-ready workflows
Streamline your mammalian and microbial clone screening projects with automated workflows. Traditionally, screening thousands to millions of cells for a target receptor, protein, or gene is labor-intensive and time-consuming. Our automation-ready systems ease this burden, significantly reducing hands-on time and creating a centralized data repository from multiple processes. Unify your lab devices to boost throughput and efficiency, minimizing human interaction and maximizing productivity.
Key features of our clone screening solutions:

Verify monoclonality with ease
Objective selection, imaging, and data collection streamline the tracking of colony formation from a single cell for confident monoclonality verification.

Efficient single-cell sorting
High-accuracy robotics and gentle fluidics systems enhance the establishment of viable clones, improving efficiency and reducing errors.

Automated workflows
Seamlessly integrate automation into your lab with end-to-end workflow solutions that provide increased throughput, consistency, and walk-away time.
Explore our portfolio of clone screening systems
QPix FLEX
Automate microbial screening from plating to picking in one compact system
The QPix® FLEX™ goes beyond colony picking, bringing together plating, streaking, liquid handling, and high-resolution color imaging into one compact, chamber-compatible system. As a fully integrated workcell, it transforms traditionally manual, error-prone steps into a seamless, automated workflow that boosts throughput, traceability, and reproducibility.

- Integrated workcell: plating, streaking, picking, liquid handling, and imaging in one system
- Compact design fits benchtop spaces and operates inside hypoxic or anaerobic chambers
- High-resolution color camera for morphology-based colony selection
- Barcode-enabled traceability for compliance and sample tracking
- Patented disposable tips eliminate cross-contamination and maintain sterility
- Ideal for microbiome, synthetic biology, and early-stage biotech applications
QPix 400 Series
Fully automate synthetic biology workflows for DNA assembly, antibody discovery and protein engineering
The QPix® 400 Series Microbial Colony Pickers combine intelligent image analysis with precise automation for fast and efficient screening of large libraries. With a variety of data tracking and assay tools, the QPix Software streamlines the control and management of complex and iterative processes.

- Automate several sample prep and plate handling processes such as transfer of bacterial liquid culture and plating on agar
- Streamline your workflow with scalable automation – pick up to 30,000 colonies per day
- Electronic data tracking for well-documented data control
- Sterile environment with customizable HEPA filtration options
ClonePix Series
Automate antibody discovery and cell line development workflows
Screen more clones with the ClonePix® Series in less time with monoclonal verification on day zero, then screen and pick for highest producers in weeks, not months.

- Screen 10X more clones than limiting dilution
- Increase probability of identifying high-value clones
- Condense the workflow into a single solution
- Eliminate or recover unstable clones early
CloneSelect Imager and CloneSelect Imager FL
Verify monoclonality confidently
The all-new CloneSelect Imager FL adds high contrast multichannel fluorescent technology in addition to the standard white light imaging that allows for accurate single-cell detection and proof of monoclonality at day 0. Streamline your workflow with comparative confluence assays to identify and verify gene edits.

- Document evidence of single cells and confluency digitally for auditing and submission to regulatory authorities
- Image cells non invasively at multiple time points to monitor colony formation
- Screen using high resolution white light imaging
- Deliver real-time results with on-the-fly analysis
- Automation and integration ready
DispenCell™ Single-Cell Dispenser
A simple single-cell dispenser for proof of monoclonality
The DispenCell Single-Cell Dispenser is an automated laboratory instrument designed for fast, easy, and gentle single-cell isolation. Cell dispensing technology enables scientists to isolate single cell lines three times faster and at a lower cost compared to existing solutions.

- Easy, intuitive setup with no cleaning or calibration required
- Software that provides instantaneous proof of clonality and traceability post cell dispensing
- Unique technology gently handles the cell sample for better viability and cloning efficiency compared to manual pipetting
- Benchtop-size designed to fit under a hood, on a bench, or in a pre-existing automated workflow
- A patented disposable tip to ensure clean isolation of single cells and no cross-contamination, which is certified free from animal products and cytotoxic material

Inscripta enables scientists to perform digital genome editing with their Onyx system integrated in a fully automated workflow that includes the QPix system

The Fred Hutch Cancer Research Center uses the ClonePix 2 system to efficiently screen and isolate positive hybridoma clones

Harvard Medical School uses the QPix colony pickers to map networks of macromolecular interactions to help understand phenotype to gene relationship

Zymergen uses the QPix colony pickers to make better microbes in industrial fermentation

University of Edinburgh uses QPix colony pickers to scale up DNA manufacturing
Applications & Research
Find an extensive collection of application notes, research, and workflows related to clone screening including cell line development, antibody discovery, and more.
Cell Line Development
Stable cell lines are widely used in a number of important applications including biologics (e.g....
Antigen / Immunogen Discovery and Optimization
Vaccine development workflows vary widely depending upon the platform (e.g. inactivated virus vs. DNA vaccine)...
Antibody Discovery Using Phage Display
Phage display is a technique used to study the interaction of proteins displayed on the surface of a bacteriophage with other...
Hybridoma Workflow for Antibody Discovery
Hybridoma technology is a method for mass-producing antibodies in a hybrid cell line generated from the fusion...
Monoclonality
Monoclonality is term that describes a cell line that originates from a single progenitor (single cell) - and is therefore monoclonal. Cell line development and assurance of monoclonality are critical steps in the process of generating biopharmaceutical molecules, such as monoclonal antibodies.
Monoclonal Antibodies (mAbs)
Monoclonal antibodies (mAbs) originate from one unique parent cell, thus binding only to a single epitope. Monoclonal antibody discovery typically refers to the screening and identification of specific antibodies that target a specific epitope for the diagnosis and treatment of diseases, like the coronavirus for COVID-19.
Synthetic Biology
Synthetic biology is a broad term that refers to the manipulation of genetic pathways to harness the power of existing biological systems in novel ways (often to manufacture molecules or proteins). Synthetic biology applies principles that are derived from engineering, specifically design-build-test-learn cycles, to biological systems. By leveraging high-throughput workflows, synthetic biologists can accelerate this process.

Lab automation for high-throughput clone screening
Mammalian and microbial clone screening projects traditionally begin with a target—a receptor, protein, or gene that participates in a biological pathway of interest. Next comes screening, where thousands to millions of cells are tested and analyzed in relation to the target. This presents a significant bottleneck for labs as it requires laborious and time-consuming methods across a wide range of analytical platforms.

Gene Editing (CRISPR/Cas9)
Gene editing is a genetic manipulation in which a living organism’s genomic DNA is deleted, inserted, replaced, or modified. Gene editing is a site-specific targeting to create breaks in DNA through various techniques and does not always involve repair mechanisms.
Related Products & Services
View our complete line of clone screening systems along with an assortment of high-performance media and labware. We also offer services for instrument customizations and robotics.

Media
An assortment of high-performance media. The use of CloneMedia ensures formation of discrete, clonal colonies with cell lines such as hybridoma, CHO, CHO-S, CHOK1 and others.

Consumables
Assortment of high-performance labware including cartridges, modules, filters and microplates to name a few.

Services
Customize your instruments, as well as automate entire workflow to meet the specific needs of your assay, method, or protocol