
Application Note
Calcium flux assay for in vitro neurotoxicity studies and drug screening
- Monitor intracellular calcium oscillations in primary neuronal cultures in real time
- Determine agents’ potencies using Peak Pro analysis algorithms
- Compare agents based on their unique effects on calcium oscillation pattern in cultures
- Analyze peak frequency, amplitude, total area under the curve, and more
Introduction
Marcela Laukova, PhD and Michael P. Shakarjian, PhD | Department of Public Health, Division of Environmental Health Science, School of Health Sciences and Practice | New York Medical College, Valhalla, NY
Cathy Olsen, PhD | Sr. Application Scientist | Molecular Devices
Neurons are electrically excitable cells in the central and peripheral nervous system that process and transmit information through electrical and chemical signals. These signals occur via synapses that allow neurons connecting to each other to form neuronal networks.
Calcium ions serve as an essential intracellular messenger in mammalian neurons, generating a multitude of intracellular signals that control key neuronal functions. In presynaptic terminals, calcium influx triggers exocytosis of neurotransmitter-containing synaptic vesicles, while postsynaptically, a transient rise of the calcium level in dendritic spines is essential for the induction of activity-dependent synaptic plasticity. Intracellular calcium signals regulate processes that operate over a wide time range, from neurotransmitter release at the microsecond scale to gene transcription, which lasts from minutes to hours.
There are multiple calcium-permeable channels in the plasma membrane (e.g. voltage-gated calcium channels, as well as ligand-operated channels, including NMDA-, AMPA- and nicotinic acetylcholine receptors), which fine tune calcium flux in neuronal cells under physiological conditions. Understanding how different receptors and their agonists alter this important regulator of neuronal function in health and disease can contribute to development of novel chemical modulators, primarily in neurotoxicity studies and in drug development.
Calcium imaging, or monitoring changes in intracellular calcium, is an extremely useful technique for investigating the variety of roles that calcium ions play in functioning neurons. Direct measurement of the dynamic calcium flux within neuronal networks reveals how neurons process signals from the extracellular space, for example, response to the addition of neurotransmitters or pharmacological agents that cause changes in intracellular calcium levels.
Here, we demonstrate the feasibility of measuring intracellular calcium flux in primary rat cortical neurons in a microplate format using the EarlyTox™ Cardiotoxicity Kit in combination with the FlexStation® 3 Multi-Mode Microplate Reader, which enables compound handling for real-time monitoring of calcium kinetics. SoftMax® Pro Software provides quantification of the signal necessary for more accurate neurotoxicity studies and, ultimately, drug development. Within SoftMax Pro Software, the Peak Pro Analysis algorithms can calculate a variety of peak attributes, including Peak Count, Peak Frequency, Peak Amplitude, Peak Width, Peak Rise Time, Peak Decay Time, and Peak Spacing (Figure 1).

Figure 1. Examples of parameters calculated by the Peak Pro Analysis algorithm in SoftMax Pro Software. Up to 14 different peak attributes can be calculated using preconfigured EarlyTox Cardiotoxicity assay protocols included in the software’s protocol library.
To demonstrate the feasibiliy of this assay, we used tetramethylenedisulfotetramine (tetramine, TETS, TMDT herein), a very potent neurotoxic rodenticide responsible for many intentional and accidental poisonings in humans. TMDT actions involve blockade of chloride ion entry through the GABAA channels, leading to unchecked depolarization, excessive influx of calcium into neurons, and excitotoxicity, resulting in seizures with refractory status epilepticus, cardiac abnormalities, coma and death. Currently no specific antidote exists and an evidence-based systematic approach to discover the best countermeasure is lacking. Therefore, we developed a valid in vitro screening method utilizing primary cortical neurons to rapidly identify potential antidotes against TMDT and other similar agents.
Here, we demonstrate that ketamine, among other drugs, effectively reversed TMDT-induced increase in the intracellular calcium and thus could prevent neurotoxic effects of this agent. These in vitro data confirmed our previous observations in a mouse model, where ketamine showed potential to rescue mice from lethal seizures and mortality induced by TMDT in a dose dependent manner (Shakarjian et al. 2012). This assay represents a reliable and cost-effective in vitro alternative to animal experimentation in the process of antidote identification but also in characterization of unknown agents involved in intracellular calcium modulation.
Methods
Preparation of primary cortical neuronal culture
Primary cortical neurons were isolated from one pregnant rat dam on gestational day 18. The dam was sacrificed according to a locally approved ethical protocol. The whole uterus was dissected and placed into a petri dish with cold phosphatebuffered saline (PBS). Embryos were removed from the uterus and transferred into another Petri dish containing cold PBS in a tissue culture hood under sterile conditions.
The heads of individual embryos were removed using small scissors and transferred to another petri dish of fresh, cold PBS. Small forceps were used to steady the head, and other forceps were used to carefully open the skull without damaging the brain tissue. The brain was then pulled away from the skull bones with partial loss of the olfactory bulbs. The cerebellum and remaining olfactory bulbs were removed from each brain.
Individual brains were transferred to sterile PBS in a petri dish. Working under a dissecting scope with magnification set to about 1.8X, each brain was oriented dorsal side up. Hemispheres were pulled slightly apart and then cut to separate them. One hemisphere was then turned so the medial (flat) side was facing up, and two forceps (#55) were used to strip the meninges from the hemisphere. One forcep was used to steady the hemisphere while the other was used to clip around the hippocampus. Finally, the brainstem was removed, leaving the cerebral cortex. The entire procedure was repeated with the other hemisphere.
All cortices were collected (optional: the hippocampi may be used for primary hippocampal culture or for other applications) and cut into approximately 1-mm pieces while still in PBS. About 2 mL of medium consisting of 1 mL of DMEM supplemented with 7.5% heat-inactivated fetal bovine serum (FBS) and 1 mL of Neurobasal Complete Medium (neurobasal medium containing 2% serum-free B27 supplement, 0.5 mM GlutaMax-I and 10 μg/mL gentamicin) was added to the minced pieces.
Three fire-polished glass Pasteur pipets with different diameters (small, medium, and large) were used to gently dissociate the tissues with 10-12 triturations per pipet, starting with the pipet with the largest opening and proceeding to the one with the smallest opening. Once tissue was completely dissociated, plain DMEM was added to the cell suspension to a total volume of about 10 mL. The suspension was then centrifuged for 5 minutes at 300g at 4°C. Medium (supernatant) was removed, and the pellet was resuspended in 1 mL of fresh Neurobasal Complete Medium.
Cells were counted using a hemocytometer and plated at 50,000– 80,000 cells per well in a total volume of 100 μL per well in a sterile 96-black wall, clear bottom well plate coated with poly-D-lysine. Cells were incubated for 1 day at 37°C and 5% CO2 to allow them to adhere. After 1 day, another 100 μL of fresh Neurobasal Complete Medium was added to each well. Cells were incubated for at least an additional 10 days, exchanging half of the medium for fresh medium every fifth day. Cells were observed daily to confirm formation of a cell monolayer, extension of neuronal processes, and neuronal network.
Calcium flux measurement
At 14 days in vitro, baseline calcium oscillations in primary neuronal culture manifest as synchronous peaks. Cell density and days in vitro are primary factors determining the frequency and amplitude of calcium peaks. This baseline calcium oscillation pattern usually changes rapidly after injection of compounds or drugs known to be involved in calcium flux, e.g. calcium channel agonists or antagonists. The FlexStation 3 reader enables up to three compound transfers in a single plate read, allowing a broad combination of compounds or drugs to be tested sequentially in a single experiment.
For dye loading of cells, 100 μL of medium was removed from each well, leaving a volume of 100 μL in each well. The EarlyTox Cardiotoxicity Kit working solution containing the calcium-sensitive dye was prepared according to the product insert, and 100 μL was added to each well. Neurons were incubated in the dark for at least 1 hour at 37°C.
Calcium flux was measured on the FlexStation 3 reader using a PeakPro Analysis protocol from the SoftMax Pro Software’s protocol library. The plate was read using Fluorescence read mode and Flex read type, which enables simultaneous detection of fluorescence signal (490 nm ex/525 nm em) from the bottom while compounds are delivered to the wells by the onboard liquid handling system. Typical settings included a baseline reading of 300 seconds, followed by a first compound addition at 300 seconds and a second compound at 800 or 1100 seconds. The total reading time in this assay is 1900 seconds. Specific settings used are shown in Table 1. Depending on the assay and drugs used, the injection times may vary. A representative kinetic trace is shown in Figure 2.
No. of compound transfers: 2
Pipette height: 70 μL, Volume: 20 μL, Rate: 2 μL/sec
Time point: 300 sec, Minimum time ~15 sec
Transfer 2:
Pipette height: 100 μL, Volume: 20 μL, Rate: 2 μL/sec
Time point: 800 sec (or 1100 sec), Minimum time ~334 sec
Table 1. FlexStation 3 reader settings.
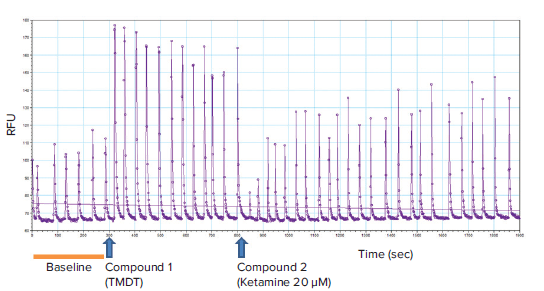
Figure 2. Representative kinetic trace with the timepoints for addition of compounds. Baseline calcium oscillation read (0 to 300 seconds) and tracings after compound addition (TMDT at 300 seconds and ketamine at 800 seconds) are indicated. Note that TMDT (10 μM) markedly increased the peak amplitude and thus the total intracellular calcium levels, as expected. The peak frequency did not appear to be significantly affected. Addition of ketamine (20 μM) reversed TMDT-induced effect upon the calcium oscillations.
In this experiment, TMDT was added to the neuronal cells after a 5-minute (300s) baseline reading. Various compounds were subsequently added to the cells to determine whether they could permanently or temporarily reverse the effects of TMDT on intracellular calcium. Representative kinetic traces for several different compounds are shown in Figure 3.
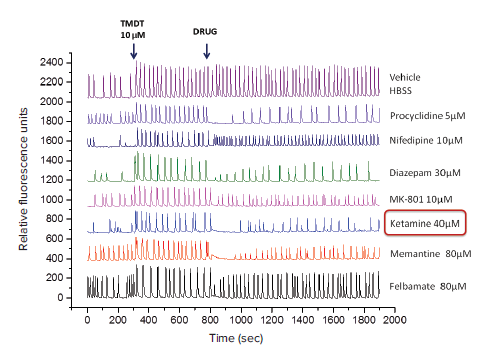
Figure 3. Representative kinetic traces using simultaneous injection of various drugs to the cells. Baseline calcium oscillation read (0 to 300 seconds), tracings after addition of TMDT at 300 seconds followed by addition of various drugs at 800 seconds) are indicated. Note that TMDT (10 μM) markedly increased the peak amplitude and thus the total intracellular calcium levels, as expected. Addition of pharmacological agents, applied at different concentrations displayed varying efficacies in reversing TMDT-induced effects upon oscillation pattern. OriginPro 2016 Software (OriginLab Corporation, MA) was used to draw all the plots in one graph.
Results
TMDT added to the neuronal cells at 10 μM reliably and significantly increased the total intracellular calcium concentration as demonstrated by AUC analysis (t-test, p<0.0001) as well as the peak amplitude (t-test, p=<0.0001), but not the peak frequency, when compared to the baseline (Fig. 4A and 4B, dotted line). Various drugs displayed differing efficacies in their ability to reverse TMDT-induced changes in the calcium oscillations (representative traces shown in Figure 3).
Ketamine used at three different concentrations reversed the effects of TMDT on the total intracellular calcium concentration (AUC, ANOVA F(3,42)=15.66, p<0.0001, Fig. 4A) primarily by decreasing the peak amplitude (ANOVA F(3,42)=13.75, p<0.0001, Fig. 4B) which showed a dose response dependence. However, the effect upon the peak frequency varied, with a significant effect being observed after addition of 40 μM ketamine (ANOVA F(3,42)=4.06, p<0.013, Fig. 4C). There was no statistically significant difference between the individual concentrations of ketamine used here.

Figure 4. Modulation of neuronal calcium flux in response to ketamine. After a baseline reading, TMDT was added to neurons using the FlexStation 3 pipettor, and calcium flux was monitored for 5min (300 s). Ketamine was then added at the concentrations indicated, and calcium flux continued to be monitored for additional 800 seconds. A preconfigured protocol using Peak Pro analysis algorithms in SoftMax Pro Software was used to calculate various parameters of the data from the raw kinetic traces. SoftMax Pro Software was used to calculate area under curve (AUC, A), peak amplitude (B), and peak frequency (C) per 100 seconds. Each parameter was expressed as an average percentage of the baseline (dotted line) ± standard error of the mean (SEM) (n=2-12). *p<0.05 compared to baseline, #p<0.05 compared to TMDT. GraphPad Software 6.07 (GraphPad Software, Inc., CA) was used to draw the graphs and run the statistics.
Conclusion
Intracellular calcium is an important mediator of many neuronal functions during physiological and pathophysiological conditions. We have demonstrated that measuring calcium flux in primary cortical neurons using the EarlyTox Cardiotoxicity Kit with the FlexStation 3 reader enables rapid screening of pharmacological agents and may help unravel their potential involvement in calcium flux within neuronal cells. Furthermore, SoftMax Pro Software enables real-time visualization of calcium kinetics and provides immediate information about the qualitative effects of experimental compounds on calcium flux, both of which are important, especially for drug screening applications. The software also automates quantitation of several parameters of the calcium signal, including area under the curve, peak amplitude, and peak frequency, and many others, providing valuable information about the mechanism of action.
Reference
- Shakarjian MP, Ali MS, Velíšková J, Stanton PK, Heck DE, Velíšek L. 2012. Differential antagonism of tetramethylenedisulfotetramineinduced seizures by agents acting at NMDA and GABA(A) receptors. Neurotoxicology. 48:100-8.