
Application Note
Accelerating Gene Edited Cell Lines with the CloneSelect Imager FL
- Easily detect successfully transfected cells
- Cut cell line and confluency tracking times
- Reject low transfection efficiency pools at an early stage
- Confirm and track CRISPR edits with multichannel fluorescence detection
- Reduce risk of overpassaging
Prathyushakrishna Macha, PhD | Research Scientist | Molecular Devices
Introduction
Gene editing and CRISPR technologies
Gene editing technology allows manipulations at the DNA level, where sequences are deleted, added, or modified using various tools. Recently many efficient strategies have been devised to edit the genome in a whole spectrum of organisms, with new methods advancing in an unprecedented fashion. Genetic modification covers a wide range of broad applications including basic research, fighting diseases, and bioproduct manufacturing. There are specific subsets of these applications including disease model development, drug development, target gene functional analyses, generation of transgenic organisms, chimeric protein production, and others. Programmable nucleases available for genome editing include zinc finger nucleases (ZFN), meganucleases, transcription activator-like effector nucleases (TALEN), and CRISPR (clustered regularly interspaced short palindromic repeats) associated protein 9 (Cas9). Targeted gene editing uses these existing technologies, but CRISPR specifically has revolutionized science by demonstrating great efficiency, ease of use, and multiplexability1,2. Nextgeneration CRISPR-Cas technologies aim not just to target DNA but also RNA with the promise of new cutting-edge tools in gene and cell therapies.
P53 gene significance
Human p53, a gene with 20kb, 11 exons, and an intron of 10kb that separates the first two exons has been a constant source of interest with a crucial role in human oncogenesis and tumor suppression. This gene has growth inhibitory properties to keep the damaged DNA in cells from replicating. This is also regulated by apoptotic activity to remove damaged cells from our system and promote normal cell proliferation. Approximately 50 percent of human cancers reported changes in the p53 gene, understanding p53 regulation provides the basis for drug/ small molecule designs. This could lead to p53 reactivation therapy in cancers and fight the p53-based chemoresistance3–8.
Cell line development challenges and our approach
The rise of gene-editing technologies, such as CRISPR/ Cas9 and prime editing, have contributed to the accelerated generation of cell models for basic and disease research. Screening of cells after gene-editing is a crucial step that is generally performed manually. It is a tedious process involving large amounts of time and hands-on effort. For an effective gene-editing process (higher transfection efficiency) multiple parameters like nucleic acid vector design (construct with target), host cell line features, and delivery method must be optimized. This could be daunting at a large scale when performed manually. An efficient workflow is essential to increase the reproducibility and scalability of gene editing, stable cell line screening, and monoclonal cell line development.
The CloneSelect® Imager FL allows screening of up to 96 or 384 transfection conditions, using confluency tracking and fluorescent channels to successfully monitor transfection through reporter gene expression detection. Multichannel fluorescent imaging significantly improves efficiency and streamlines the workflow, generating a cell pool and eventually a monoclonal cell line with day zero monoclonality tracking. The CloneSelect Imager FL system effectively images cells grown (suspension or adherent) and transfected using fluorescent channels. These images are analyzed to select the highest transfection efficiency and protein of interest expression levels. The CloneSelect Imager FL system has two fluorescent channels, plus white light for total confluency detection.
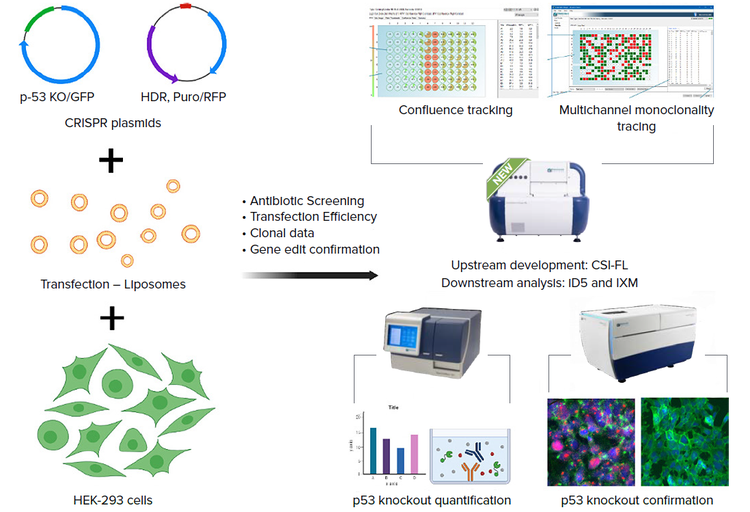
Representation of STEPS followed for CRISPR-based gene editing and verification
Here we have selected HEK-293 as our host cell line and p53 knockout (KO) system to generate a CRISPRedited pool. These were closely monitored to generate a gene-modified pool. p53 is known to upregulate growth arrest, and apoptosis-related responses to stress, therefore leading to programmed cell death, cell cycle, and differentiation changes. The edited cells were then tagged with an antibody for p53 KO confirmation, followed by apoptosis assay to compare the effects of p53 knockout in modified vs. wildtype HEK-293 cells.
Methods
Cell culture and transfection
HEK-293 cells from ATCC were maintained as per instructions before they underwent TP53 knockout using the Santa Cruz p53 CRISPR/Cas9 KO plasmid and p53 Homology-directed repair (HDR) plasmid following the manufacturer’s instructions (Santa Cruz Biotechnology). The cells were seeded at 50k/well-seeding density in a 96 (Corning 3300) well plate, cells were about 60% confluent on the day of transfection in serum-free media as per instructions provided with the TransIT-X2 delivery system (Mirus Bio). CRISPR/Cas9 TP53 KO plasmid (sc-416469) and the HDR plasmid (sc-416469-HDR; Santa Cruz Biotechnology) were co-transfected with the select reagent in different ratios (1:2, 1:3), and 24h post-transfection cells were selected with puromycin.
Antibiotic selection with CloneSelect Imager FL
The puromycin concentration was 1.5 µg/mL for the first 72 hours and then gradually tapered to 0.5 µg/mL through media change for regular maintenance until stable line generation. The cells were regularly monitored before and after puromycin selection on CloneSelect Imager FL multichannel confluence run. The cells were tracked, media was changed every 2 to 3 days, and cells were moved to 48-well tissue culture-treated plates, and then to 6-well plates for following assays.
Immunostaining
Once expanded, the cells were grown in tissue culturetreated 96-well plates until confluency and fixed with 4% paraformaldehyde and washed in three changes of PBS. These cells were labeled with p53 Antibody (DO-1) conjugated to Alexa Flour 647 (sc-126 AF647, Santa Cruz Biotechnology), Phalloidin 488(1:400), Hoechst 3342(1:1000) with 3% Blocking reagent (BSA) for overnight and washed in three changes of PBS. The images were acquired using the 10X objective of the ImageXpress® Micro Confocal High-Content Imaging System (IXM-C) from Molecular Devices.
Annexin V
48 hours after seeding, a 96-well plate of HEK-293 cells was exposed to staurosporine (5 μM) in 1:2 serial dilutions for four hours to induce apoptosis. Apoptosis was then detected with the Annexin V-FITC Apoptosis kit (ab14085) according to the manufacturer’s instructions. Images were acquired using the 10X objective of the IXM-C, and the results were analyzed using MetaXpress® and SoftMax® Pro software.
Results and Discussion
To target p53 gene knockout in HEK-293 via CRISPR/Cas9 system, we selected two vectors – one for targeting and one for antibiotic selection – and transiently transfected using a lipid-based system. The transfection efficiency was higher for cells transfected with a 1:3 ratio of DNA to transfection reagent. The efficiency was higher with the increase in transfection reagent, but this ratio was not increased further to avoid any potential cellular toxicity. The cells successfully transfected with p53 KO plasmid fluoresced in green (GFP) and the ones with both p53 KO and HDR in red (RFP) (refer to Figure 3 A, B). The rapid estimation of transfection efficiency and the screening with puromycin was carried out using the CloneSelect Imager FL (Figure 1, 2, and 3). These cells were further selected to obtain a stable pool of p53 KO cells (Figure 3- top).
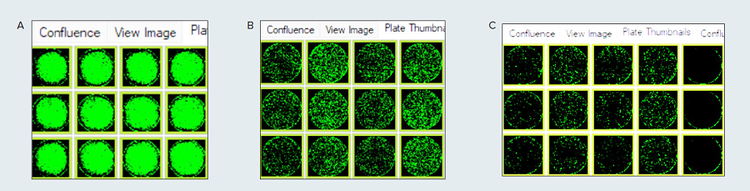
Figure 1. Plate thumbnails of HEK-293 cells in puromycin selection media, post-transfection with CRISPR-based plasmids for p53 knockout. Reporter signal and confluency measurement were tracked using CloneSelect Imager FL for further expansion. A. White light imaging of the cells and confluence representation of all living cells (pseudo green highlights applied via software) B. GFP imaging, represents successfully transfected with p53 KO plasmid with GFP reporter (confluency in pseudo green) C. RFP imaging, represents successfully transfected with HDR plasmid with RFP reporter (confluency in pseudo green)
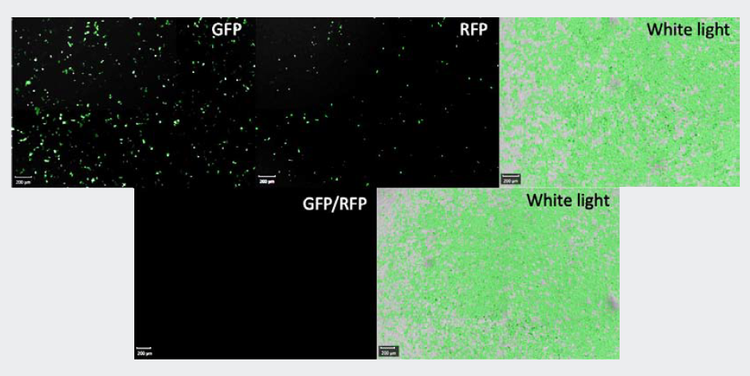
Figure 2. A1 (top) and A5 (bottom) well images in GFP (left), RFP (middle), and white light filters (right) (pseudo green cell confluence generator). A1 was transfected with both CRISPR-based plasmids for the knockout and antibiotic resistance whereas the well A5 was not, hence the absence of fluorescent protein-based reporting in cell distribution (bottom left).

Figure 3. Cell confluence and cell number estimation for each cell displayed during puromycin selection of transfected HEK-293 cells and control cells. The distribution of cells is estimated by the software for TL, GFP, and RFP imaging (top number panel). ImageXpress images of cells after transfection before puromycin screening (bottom image panel). A. TL image of transfected HEK-293 (all cells), B. FITC image (GFP expressing cells), C. Texas Red image (RFP expressing cells), and D. Overlay of B and C (expressing both GFP and RFP).
The pool generated was further selected with puromycin for better growth and scaled up for further assays. Antibody staining for p53 when performed on the successfully screened and control HEK-293 cells indicated an absence of p53 expression (Figure 4B), whereas the presence of red stain in control cells indicated the presence of p53 expression (Figure 4A). The successful knockout of p53 was further verified with double staining based on the Apoptotic Annexin V assay. There was a significant difference in apoptosis rate between the KO and control HEK-293 cells. Since p53 has a crucial role in apoptosis, the p53 knockout led to significantly lower apoptotic activity (Figure 5) induced with staurosporine treatment. The graphs in red correspond to control wells and the blue to the p53 knockout wells (Figure 5), respectively. There was almost a 10-fold difference between edited and control cells at the the highest concentration of staurosporine (5 µM). Though the fold difference reduced with a drop in drug concentration the control cells still had an overall higher apoptotic activity.
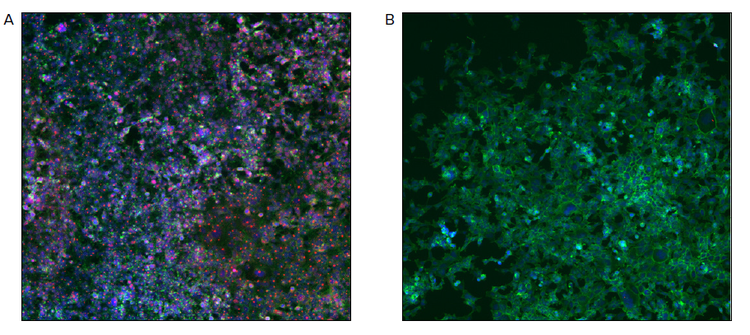
Figure 4. . Composite images of p53 KO HEK-293 cells (B) and control cells (A) labeled with p53 Antibody (DO-1) conjugated to Alexa Flour 647 (sc-126 AF647, Santa Cruz Biotechnology), Phalloidin 488(1:400), Hoechst 3342(1:1000).
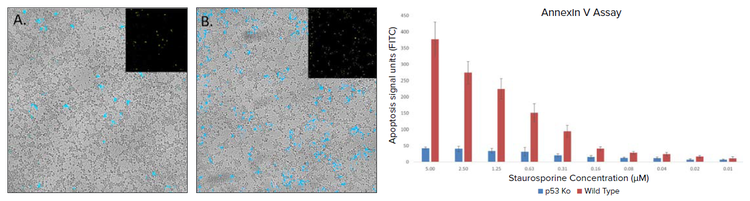
Figure 5. Cell apoptosis detected by Annexin V-FITC (Top image panel), overlay of white light, and FITC Images. A. p53 KO cells treated with Staurosporine (2.5 μM) and B. Control cells treated with Staurosporine (2.5 μM) . Graphical representation of data obtained using ImageXpress and analyzed by MetaXpress software (bottom). FITC signal indicating apoptotic activity in the treated cells is plotted against the concentration of staurosporine.
Conclusion
The experimental study established an effective and rapid process for developing a p53 knockout HEK-293 cell pool using CRISPR-based gene editing and antibiotic screening. This was effectively carried out using our CloneSelect Imager FL. The cells were further studied using various imaging assays to assess the knockout of p53. The knockout has decreased the average apoptotic activity of the cell pool substantially on induction of apoptosis. CloneSelect Imager FL has enabled a more efficient screening of gene-edited cells through fluorescence detection.
References
- Maeder, M. L., & Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Molecular Therapy, 24(3), 430-446.
- Khalil, A. M. (2020). The genome editing revolution. Journal of Genetic Engineering and Biotechnology, 18(1), 1-16.
- Haupt, S., Berger, M., Goldberg, Z., & Haupt, Y. (2003). Apoptosis-the p53 network. Journal of cell science, 116(20), 4077-4085.
- Amaral, J. D., Xavier, J. M., Steer, C. J., & Rodrigues, C. M. (2010). The role of p53 in apoptosis. Discovery medicine, 9(45), 145-152.
- Brown, C. J., Lain, S., Verma, C. S., Fersht, A. R., & Lane, D. P. (2009). Awakening guardian angels: drugging the p53 pathway. Nature Reviews Cancer, 9(12), 862-873.
- Liu, Y., Wang, X., Wang, G., Yang, Y., Yuan, Y., & Ouyang, L. (2019). The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. European journal of medicinal chemistry, 176, 92-104.
- Cao, X., Hou, J., An, Q., Assaraf, Y. G., & Wang, X. (2020). Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resistance Updates, 49, 100671.
- Stiewe, T., & Haran, T. E. (2018). How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resistance Updates, 38, 27-43.
- Pickar-Oliver, A., & Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nature reviews Molecular cell biology, 20(8), 490-507.
- Gao, P., Lyu, Q., Ghanam, A. R., Lazzarotto, C. R., Newby, G. A., Zhang, W., ... & Miano, J. M. (2021). Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome biology, 22(1), 1-21.